All potential drugs and environmental chemicals produced in large volumes are tested for toxicity using animals. The tests have changed little in several decades, and are now known to be inaccurate. Could it be because the experiments are poorly designed? In particular, by using a single stock of mice or rats, they fail to take account of genetic variation in response. If they happen to choose a resistant stock, toxicity may be completely missed, and if it is genetically heterogeneous statistical power will be reduced. This is explored in detail in this page.
Inadequacy of current toxicological methods
Properties of “genetically defined†isogenic strains and “genetically undefined†outbred stocks
Examples showing strain differences in response to xenobiotics and demonstrating how a multi-strain test could reduce variability and uncertainty
Example 1: the response of two strains of rats to diethylstilbestrol (DES)
Example 2: Use of small numbers of animals of several strains would be a better strategy in carcinogenesis testing (and in other applications)
Example 3: shows how it is possible to use several strains without increasing total numbers
Example 4: shows that short-term multi-strain assays are practical and powerful
Example 5: multi-strain versus outbred stock toxicity test of the effects of chloramphenicol
Example 6: shows how use of genomic techniques (microarrays) can explain strain differences in response to a carcinogen
Discussion
Conclusions
References
Inadequacy of current toxicological methods
According to the FDA “Critical Path†White Paper, 2004,
( http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html)
“The traditional tools used to assess product safety — animal toxicology and outcomes from human studies — have changed little over many decades and have largely not benefited from recent gains in scientific knowledge. The inability to better assess and predict product safety leads to failures during clinical development and, occasionally, after marketing.â€
The most noticeable advances in recent years have been the sequencing of the human, mouse and rat genomes and the development of molecular techniques sometimes referred to as the “omicsâ€.
- The sequence of many thousands of genes is known, and the expressions of individual genes can be measured using gene microarrays.
- Mapping and identification of quantitative trait loci is now possible, and is becoming increasingly easy
- Mice and rats can be genetically modified so that the normal and abnormal action of individual genes and their interaction with the rest of the genome may be studied.
- The genomes of the mouse and rat are turning out to be remarkable similar to humans, and even many genetic linkages have been conserved. As this new information accumulates, maintained by the relatively new discipline of informatics, genetically defined animal models become increasingly valuable in gaining an understanding of human disease.
However, toxicologists are not benefiting from these major advances. One important reason for this is that they use the wrong animals. Most toxicity testing of pharmaceuticals is done using genetically undefined Sprague-Dawley or Wistar rats and CD-1 or “Swiss†mice.
- Each experiment is a “one-off†using genetically different animals each time.
- No useful genomic information is generated because it is impossible to tell whether any observed variation is due to genetic or non genetic causes, and because the genotype of each animal is unknown. So useful information does not accumulate.
- And because toxicologists only ever use a single strain, they are rarely aware of strain differences which reveal genetic variation in response.
Strain differences in response to xenobiotics (20) can be very marked. For example, the LD50 of TCDD varies almost a thousand-fold between different strains of rats(21). Such genetic variation in the test animals needs to be taken into account in routine toxicity testing, but possibly more importantly, it provides additional information on mechanisms and possible human responses to the test chemicals. For many years geneticists have been pointing out that a better strategy would be to use a small battery of isogenic strains as the test population, without increasing the total numbers of animals which are used. This would increase the power of the experiments, would reduce variation and uncertainty, would not substantially increase costs and would facilitate the use of new scientific methods collectively designated the “omicsâ€. (2;2;4-17).
Properties of “genetically defined†isogenic strains and “genetically undefined†outbred stocks
Inbred strains of mice and rats are now nearly 100 years old, and for many decades geneticists have been advocating their more widespread use. For example, in 1936 Dr. C.C. Little stated (22):
“Just as the purity of the chemical assures the pharmacist of the proper filling of the doctor’s prescription, so the purity of the mouse stock can assure a research scientist of a true and sure experiment…….In experimental medicine today….the use of in-bred genetic material…is just as necessary as the use of aseptic and anti-septic precautions in surgery”
The properties of inbred strains can be considered under the following headings:
Isogenicity. Every animal within an inbred strain (and F1 hybrid between two inbred strains) is genetically identical. This means that treated and control animals used in an experiment will be genetically identical, which is not true with a genetically heterogeneous outbred stock.
An individual can be genotyped at a particular locus in the knowledge that all other animals of that strain will have the same genotype. Once this has been done, the data is available to all research workers using that strain. In contrast, the genotype of an outbred individual, such as a Sprague-Dawley rat, is never known unless it is individually typed. Already the full DNA sequence of fifteen inbred strains of mice has been determined. This will provide invaluable data in designing and interpreting experiments using these strains. It would not be sensible to sequence the DNA of outbred mice or rats as every animal has a different sequence.
Homozygosity. Each individual of an inbred strain is homozygous at virtually all loci. This means that they will breed true and will not carry hidden recessive genes. F1 hybrids will be heterozygous at all loci at which the parental strains differ, so these will not breed true. The level of heterozygosity of outbred stocks will vary, depending on the previous history of the colony. Colonies maintained in small numbers for many generations will be relatively homozygous. Outbred stocks can carry recessive deleterious genes which may complicate the interpretation of some experiments.
Phenotypic uniformty. There is no genetic variation within an inbred strain of F1 hybrid. However, there is some non-genetic variation. Each animal has a slightly different environment, for some characters there may be measurement error, and some characters, such as the development of a tumour, may depend partly on chance.
- So inbred strains and F1 hybrids are more uniform than outbred stocks, at least for characters which are highly inherited.
- This means that experiments using such animals will either be more powerful than ones using outbred stocks, or sample size can be reduced. This is illustrated in Example 4, below. Phenotypic uniformity is of particular importance when using genomic techniques such as gene microarrays. These are expensive techniques, so sample size must be kept small. Lack of genetic variation within each strain is of particular importance in such work.
Long-term stability. Inbred strains can only change as a result of new mutations. Although these are relatively rare, over a period of several generations some of these will become fixed and the strain characteristics will gradually change. This can be prevented by maintaining banks of frozen embryos. Selective breeding and further inbreeding will not change strain characteristics. Because of this stability it is possible to build up a database of the characteristics of each strain such as lifespan, tumour incidence, spontaneous diseases, behaviour and response to xenobiotics.
In contrast, outbred stocks can change quite rapidly as a result of changes in gene frequency due to genetic selection and inbreeding. Although this could be reduced by embryo freezing, it is not so practical because large numbers of embryos need to be stored in order to preserve all the genes present in the population. It is simply not worthwhile phenotyping outbred stocks to any great extent because of theirlability.
Individuality. Each inbred strain is unique and different from every other strain. It may be susceptible to certain inherited diseases, microorganisms, or xenobiotics. Because of strain differences it is essential that screening experiments are replicated across several strains in order to ensure a reasonably full range of responses. This point is illustrated in Examples 1, 2, 4 and 5. Examples 2, 3, 4 and 5 illustrate how it is possible to use several strains without increasing the total number of animals.
International distribution. Because inbred strains are isogenic, a single breeding pair carries all the genes present in the strain. Breeding pairs can be distributed to laboratories throughout the world in the knowledge that all scientists can be working on the same animals. Their findings are published, and become part of the historic record of strain characteristics. In contrast, every colony of outbred Sprague-Dawley rats is different and each research project using such outbred stocks uses genetically different animals. The historical record is therefore completely unreliable, even though two stocks may have the same name.
Identifiability. The authenticity of an inbred strain can easily be tested using a range of DNA markers. So genetic quality control is relatively easy. So long as a source of DNA is available the genotype of any individual can be verified.
However, genetic quality control of outbred stocks is so much more complicated that it is rarely attempted.
- There is not even a reliable set of genetic markers which will distinguish genetically between Sprague-Dawley and Wistar rats or between different outbred stocks of mice.
- Genetic quality control of outbred stocks is not easy. It is necessary to use large sample sizes and test many individual loci to ensure that gene frequency has not changed with respect to some previous standard. If drift is detected, it would then be difficult to know what to do about it.
Sensitivity. There is some suggestion that inbred strains are more sensitive, and less robust than outbred stocks. Certainly they have a poorer breeding performance. This may have adverse consequences for teratology and multi-generation experiments, and those involving neonates. It is possible to use F1 hybrids for such studies, but their offspring will not be isogenic. A relatively prolific inbred strain mated to a different strain could be used for teratology studies. The offspring would then be F1 hybrids. Their extra vigor means that litter size is somewhat larger than if they were inbred. However, the use of inbred strains in reproductive studies does require further consideration.
Examples showing strain differences in response to xenobiotics and demonstrating how a multi-strain test could reduce variability and uncertainty
Toxic end-points such as death or presence of a tumour are dependent on the sensitivity of the strains which are used. Figure 1 shows the response of two strains of rats to diethylstilbestrol(23). In the outbred Sprague-Dawley rats there was a low incidence of spontaneous mammary tumours (this was the only significant type), but this was reduced in the treated group. Exactly the opposite was found in the ACI strain rats in which the treated group had over 70% tumours. Four conclusions can be drawn from this slide:
- The outcome of a conventional toxicity test can depend entirely on the strain of animals which are used. There is no a priori way of choosing a sensitive strain.
- A toxicologist using Sprague-Dawley rats alone would have no way of knowing that the response was under genetic control, and would erroneously conclude that DES is not carcinogenic in rats. DES was given to many pregnant women in the late 1950s with their daughters subsequently getting vaginal cancer. If it was tested on SD rats then it would have appeared to be quite safe.
- Toxicologists mostly use genetically heterogeneous rats and mice on the grounds that
“..it is more correct to test on a random-bred stock on the grounds that it is more likely that at least a few individuals will respond to the administration of an active agent in a group which is genetically heterogeneous”(1).
Clearly, this does not work. There were no Sprague-Dawley rats which responded to DES by producing tumours. There are good genetic reasons for this. Much of the genetic variation is unexpressed because many animals are heterozygous for recessive sensitivity genes, there may be substantial epistasis (the effect of one gene prevents the expression of other genes. For example, albinism hides other coat colour genes), and sample sizes are small.
- An experiment involving half the number of animals of each of two strains would easily have shown that DES causes cancer in rats.
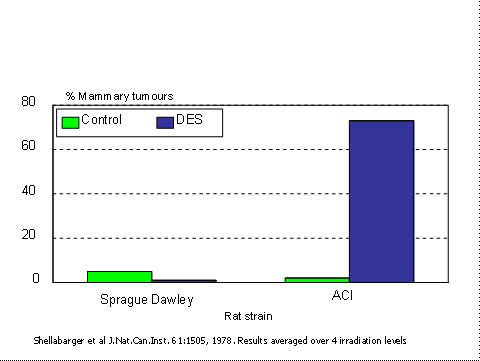 Figure 1. Response to diethylstilbestrol in two strain of rats. Note “n†is greater than 90 in each case. Data averaged over radiation levels.
Figure 1. Response to diethylstilbestrol in two strain of rats. Note “n†is greater than 90 in each case. Data averaged over radiation levels.
Example 2. Use of small numbers of animals of several strains would be a better strategy in carcinogenesis testing (and in other applications)
Figure 2 shows strain differences in susceptibility to the development of prostate tumours following treatment with another carcinogen(24). Note that the Wistar stock was entirely resistant, whereas F344 was susceptible with nearly 50% of rats getting tumours.
Typically a carcinogenesis bioassay uses a single strain of animals with four groups of 50 rats per group. If the above figure was a true representation of the range of susceptibility of rat strains, then there would be a 1/5 chance of getting a false negative result. It could be higher. The 7% tumours found in the CD stock could even be missed as that represents only three or four tumours in a sample of 50 rats.
Had the test population consisted of ten rats of each of the five strains, then the incidence of tumours would have been 21% which would be easily detectable, and there would have been no false negative results. The statistical implications of using several different strains in a carcinogenesis screen, without increasing total numbers have been considered in detail (3;19), with the conclusion that the multi-strain design is more powerful than the use of a single strain except in the rare circumstance where susceptibility is rare and the most sensitive strain happens to be chosen. These studies also suggest that the more strains that are used, the more powerful the experiment becomes. However, there are practical limitations on the number of strains which might be used.
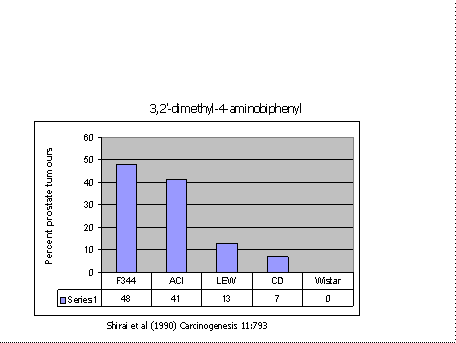 Figure 2. Percentage of rats of five strains getting prostate tumours following treatment with 3,2′-dimethyl-4-aminobiphenyl
Figure 2. Percentage of rats of five strains getting prostate tumours following treatment with 3,2′-dimethyl-4-aminobiphenyl
Example 3 shows how it is possible to use several strains without increasing total numbers
- Many toxicologists assume that if two strains are to be used this will require twice as many animals.
- In fact, there is no need to use any more animals. Table 1 shows that if the aim is to compare eight control and eight treated rats for their response to a potentially toxic agent it would be possible to use two animals of each of eight inbred strains.
- This experiment is the genetic equivalent of a human study using monozygous twins. It should be more powerful than an equivalent experiment using outbred stocks because the treated and control group would be exactly matched. Assuming a quantitative (measurement) outcome, the experiment would be analysed using a paired t-test rather than the two-sample t-test which would be used with an outbred stock.
- Although this is a perfectly valid design, and would usually be more powerful than the use of an outbred stock it would normally be better to have at least two animals of each strain in the control and treated groups as this provides for a statistical test of whether strains differ in response. Thus this experiment could be improved by using four strains with two animals of each strain per treatment group at the cost of having a slightly narrower range of genotypes being tested..
| Table 1. A hypothetical experiment using eight inbred rat strains to compare treated and control groups | |
| Control | Treated |
| LEW | LEW |
| F344 | F344 |
| DA | DA |
| WKY | WKY |
| ACI | ACI |
| BDX | BDX |
| BN | BN |
| PVG | PVG |
Example 4 shows that short-term multi-strain assays are practical and powerful
Three parallel experiments on the toxicity of gentamicin were done by PhysioGenix, as small company in Wisconsin, USA. The rats received 240 mg/kg/day i.p for six days:
- Group 1 involved 16 control and 30 treated Sprague-Dawley rats
- Group 2 involved 15 control and 31 treated F344 inbred rats
- Group 3 consisted of seven F1 hybrid strains (2-4 rats per strain, 25 control and 21 treated) of rats
- A total of 34 outcome parameters were measured (body weight at five time points, left and right kidney weight, heart weight and 26 biochemical characters).
- Data has been expressed as the absolute difference between the treated and control group means (i.e. the response) divided by the within-group standard deviation. Thus response is expressed in standard deviation units (directly related to Student’s t) for all 34 traits are shown in Fig. 3.
In general the same characters were up or down-regulated in all three sets of data, but the responses were generally much larger in the isogenic studies, leading to more powerful experiments.
- The two horizontal lines show the effect size that a future experiment using 23 rats per group would be expected to be able to detect assuming a 5% significance level, a 90% power, and a 2-sided Student’s t-test.
- About five significant differences among the 34 traits are likely to be detected when using Sprague-Dawley rats,
- About 15 significant differences when using F344 and
- About 18 differences when using the multi-strain design. With the multi-strain design about five significant differences would be likely to be detected using only 4 rats per group (not shown). Thus in theory, with the multi-strain design, it would be possible to decrease sample size quite a lot and still increase statistical power.
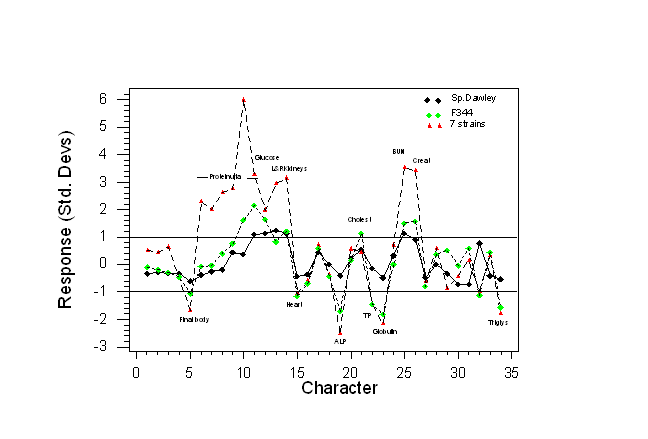 Fig 3. Absolute response in standard deviation units for 34 characters in three experiments involving Sprague-Dawley rats, F344 rats or 7 strains. Each experiment involved a total of 46 rats (Data from Dr. Howard Jacob, Physiogenix, Wisconsin, USA).
Fig 3. Absolute response in standard deviation units for 34 characters in three experiments involving Sprague-Dawley rats, F344 rats or 7 strains. Each experiment involved a total of 46 rats (Data from Dr. Howard Jacob, Physiogenix, Wisconsin, USA).
The horizontal lines at +/- one Std. Dev. indicate the magnitude of response in some future experiment that would be expected to be detectable with a 90% probability, a 5% significance level and a sample size of 23 animals per group using a two-sided t-test.
Note that with the Sprague-Dawley rats (black diamonds) only differences from the controls for proteinuria on the last day, blood glucose, left and right kidneys and BUN (5 characters) lie outside the horizontal lines, so would be likely to be detectable. For the 7-strain design about 16-17 characters in the treated rats would be likely to be different from the controls. Clearly, the 7-strain design is much more sensitive to the toxic effects of gentamycin.
Example 5 multi-strain versus outbred stock toxicity test of the effects of chloramphenicol
In humans chloramphenicol can cause a dose-dependent, reversible mild anaemia and leukocytosis. It can also cause pernicious anaemia which can be fatal, but this can not be modeled in laboratory animals.
- The haematological response of CD-1 mice (8/group) to chloramphenicol at six dose levels is shown in Fig. 4a
- The same haematological response to chloramphenicol of a multi-strain study using two mice of each of four inbred strains (BALB/c, C57BL/6, CBA and C3H, a total of 8 mice/group), averaged across strains is shown in Fig. 4b.
- The responses have been expressed as the treatment mean minus the control mean divided by the within-group standard deviation. Data comes from Ref. 16.
Results:
- In Fig. 4a a reticulocyte response is shown at a dose of 2000mg/kg, and a haemoglobin and platelet response at the highest dose level.
- In the multi-strain study responses in reticulocytes, platelets, haematocrit and haemoglobin are seen at the 1500mg/kg dose level and white blood cell responses are seen at the highest dose levels.
- Thus in this case there was a qualitative difference in response between the two designs, with the multi-strain experiment replicating the lymphocytopenia seen in humans.
- There was also a statistically significant (p=0.007) strain x treatment interaction for the LYMPH response, with BALB/c being resistant like CD-1 and the other three strains being susceptible, like the human response (not shown).
- The average response (difference between mean of treated and control group) averaged across all dose levels and characters was 0.694 standard deviations in the CD-1 mice and 1.534 standard deviations in the 4-strain study. Suppose a scientist wanted to set up a study to detect a response of these magnitudes in a two group test (control versus treated) with a significance level of 0.05, a two-sided test and a power of 90%, a power analysis shows that it would be necessary to use 45 CD-1 mice per group or 10 inbred mice (2-3/strain although in practice it might be better to use 12 mice per group with 3 mice of each strain). So the same conclusions could be reached using approximately a quarter of the number of inbred than outbred mice.
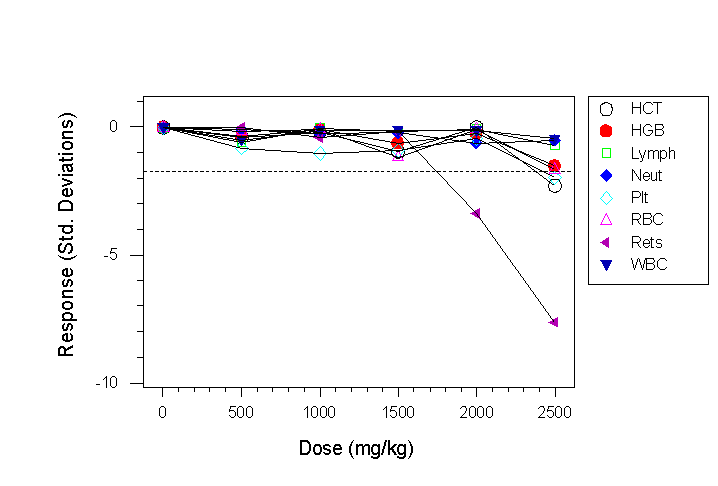 Fig 4a. Haematological responses to chloramphenicol in CD-1 mice. The dotted line in this case shows the response that should be detectable in a future experiment assuming a sample size of 8 mice/group, a 90% power, a 5% significance level and a two sided t-test.
Fig 4a. Haematological responses to chloramphenicol in CD-1 mice. The dotted line in this case shows the response that should be detectable in a future experiment assuming a sample size of 8 mice/group, a 90% power, a 5% significance level and a two sided t-test.
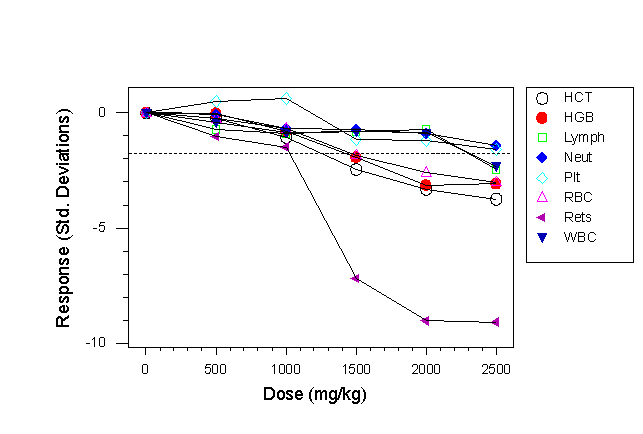 Fig 4b. Haematological response to chloramphenicol in a multi-strain study using four inbred strains (C57BL/6, CBA/Ca, C3H/He, BALB/c). Individual strain responses are not shown.
Fig 4b. Haematological response to chloramphenicol in a multi-strain study using four inbred strains (C57BL/6, CBA/Ca, C3H/He, BALB/c). Individual strain responses are not shown.
One of the most significant advantages of the multi-strain design is that it opens up the possibility of using recent advances in genomics in toxicity tests. As an example, many carcinogens, such as urethane, cause lung cancer in strain A/J mice, whereas strain C57BL/6 is resistant. Genetic mapping shows that susceptibility has a polygenic mode of inheritance with two major loci situated close to Kras2 on chromosome 6 and the H2 complex (the MHC) on chromosome 17 (18). Urethane is all metabolised within the first 24 hours, but the tumours do not appear until about 5 months after exposure. Fig. 5a shows gene expression in the lungs of mice 6-48 hrs after exposure to urethane (unpublished work of Yang et al).
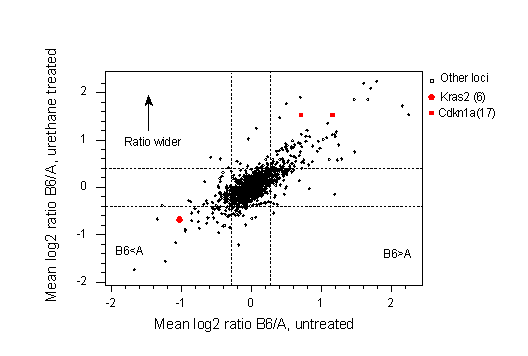 Fig 5a. cDNA expression of 1389 genes in the lungs of mice after exposure of urethane using a cDNA microarray. The X-axis shows the relative expression in untreated A/J and C67BL/6 mice averaged across three microarrays 6, 24 and 48 hrs. after treatment. The Kras2 and Cdkn1a loci, mapping to chromosome 6 and 17, respectively, are shown in red. As this is a log2 scale, it shows that the oncogene Kras2 is 2-fold over expressed in strain A/J compared with strain C57BL/6 whereas the tumour suppressor gene Cdkn1a is approximately 2-fold over expressed in C57BL/6. The Y-axis shows the ratio of cDNA in the lungs of mice treated with urethane. Dotted lines represent the 95% confidence interval for a ratio of zero. On the X-axis genes t the left of the left-hand dotted line are significantly over-expressed in A/J relative to B6, with the converse being true with the right hand dotted line.
Fig 5a. cDNA expression of 1389 genes in the lungs of mice after exposure of urethane using a cDNA microarray. The X-axis shows the relative expression in untreated A/J and C67BL/6 mice averaged across three microarrays 6, 24 and 48 hrs. after treatment. The Kras2 and Cdkn1a loci, mapping to chromosome 6 and 17, respectively, are shown in red. As this is a log2 scale, it shows that the oncogene Kras2 is 2-fold over expressed in strain A/J compared with strain C57BL/6 whereas the tumour suppressor gene Cdkn1a is approximately 2-fold over expressed in C57BL/6. The Y-axis shows the ratio of cDNA in the lungs of mice treated with urethane. Dotted lines represent the 95% confidence interval for a ratio of zero. On the X-axis genes t the left of the left-hand dotted line are significantly over-expressed in A/J relative to B6, with the converse being true with the right hand dotted line.
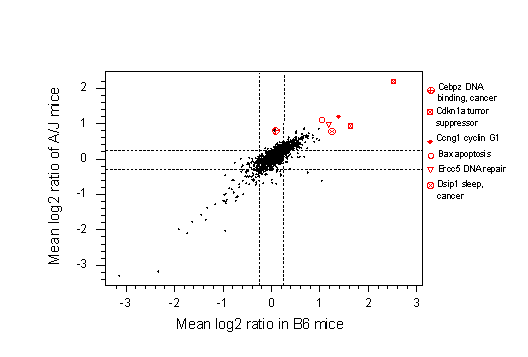 Fig. 5b. This shows gene expression in the lungs of urethane-treated relative to untreated C57BL/6 mice on the X-axis and on the Y-axis a similar ratio for strain A/J. Results are averaged across 12 microarrays, three each at 6, 24 and 48hrs. post treatment. Note that the most up-regulated genes, shown with individual symbols, are concerned with DNA repair (Ercc5), DNA binding (Cebpz), apoptosis (Bax) and the cell cycle (Cdkn1a). Dsip (delta sleep-inducing protein) may be up-regulated because urethane is an anaesthetic, although it is also cancer-associated. Thus, although strain C57BL/6 is resistant to the carcinogenic action of urethane it still responded to urethane as a carcinogen. The Cdkn1a locus was significantly up-regulated at 6 hrs in C57BL/6, but not in A/J, a difference that was statistically significant (p<0.05, not shown).
Fig. 5b. This shows gene expression in the lungs of urethane-treated relative to untreated C57BL/6 mice on the X-axis and on the Y-axis a similar ratio for strain A/J. Results are averaged across 12 microarrays, three each at 6, 24 and 48hrs. post treatment. Note that the most up-regulated genes, shown with individual symbols, are concerned with DNA repair (Ercc5), DNA binding (Cebpz), apoptosis (Bax) and the cell cycle (Cdkn1a). Dsip (delta sleep-inducing protein) may be up-regulated because urethane is an anaesthetic, although it is also cancer-associated. Thus, although strain C57BL/6 is resistant to the carcinogenic action of urethane it still responded to urethane as a carcinogen. The Cdkn1a locus was significantly up-regulated at 6 hrs in C57BL/6, but not in A/J, a difference that was statistically significant (p<0.05, not shown).
These two microarrays provide a plausible explanation of why strain A/J mice get lung tumours, independent of the carcinogen, whereas C57BL/6 mice are resistant. Compared with C57BL/6 mice, untreated A/J mice have a 2-fold elevated level of Kras2, an oncogene, and a 2-fold lower level of Cdkn1a, a tumour suppressor. Moreover, Cdkn1a is more than 2-fold up-regulated in C57BL/6 at six hours post treatment, but in A/J this level of up-regulation is only seen at 24hrs (not shown).
Discussion
For more than three decades geneticists have published papers both in toxicology journals and in more general ones such as Nature(6) and Nature Genetics(2) suggesting that toxicity tests should be done using a small battery of isogenic strains rather than the single outbred stock or inbred strain as currently used. At no time have toxicologists attempted to defend current methods. They have simply ignored the criticisms. This was understandable because of the difficulty of negotiating an entirely new design of study with regulators who were assumed to be extremely conservative. But if the FDA is dissatisfied with current methods, then new methodology should certainly be considered.
One problem is that many toxicologists believe that the use of a single outbred stock is appropriate because it more closely resembles the genetically heterogeneous human population. For example, in a response to this web site, one anonymous toxicologists stated:
“The variability of toxicity obtained in less well defined animals is a strength in itself, not a problem, when trying to predict safety margin in the non-isogenic human population.â€
However, rats and mice are not little humans. It is a fundamental assumption that if a compound is toxic in rats and/or mice it may be toxic in an approximately similar manner in humans. Thus the aim of the toxicity test is to find out in what way and at what level the compound is toxic in rats and mice. Introducing uncontrolled phenotypic variation only reduces the statistical power of the test, leading to false negative results, and in no way improves extrapolation to humans. In fact toxicologists specifically avoid such variation by using age and weight matched animals. Moreover, Example 1 shows clearly that such variation can not be relied on to detect toxicity, and nor can it be used to identify genetic variation in response. The multi-strain assay overcomes both these problems.
A second common objection is because some toxicologists do not understand the concept of the factorial experimental design. Currently a carcinogenesis assay uses 50 mice or rats per group, usually with 4 dose levels and two sexes. A multi-strain carcinogenesis study could use, say, 10 rats of each of five strains instead of 50 rats of one strain. Tumour incidence is then averaged across the five strains. However, many toxicologists do not see this as a single experiment. They mistakenly regard it as five separate experiments, each of which is too small. Of course most of them also use outbred animals. If each treatment by genotype group is to be considered a separate experiment, then by this reasoning they actually have 50 separate experiments each of a single rat, but because they can not identify the genotype of individual animals, they treat them as if they are all the same. Factorial designs have been used for well over 70 years and are discussed in detail in most statistical texts. If toxicologists do not understand the concepts, then it is the fault of their lack of training in statistical methods.
A further objection is that inbred strains are all in some way abnormal. Some strains have been developed for a high incidence of tumours, they are valuable in cancer research but may not be suitable for testing carcinogens. Other strains develop specific diseases such as hypertension or diabetes. Again these strains may not be suitable for toxicological screening. However, there are many long-lived strains which are sensitive to toxic agents and do not develop such specific diseases which would appear to be suitable for toxicological screening. Where a specific disease model is wanted, then sometimes it is available as a inbred strain or as a genetically modified strain on an inbred genetic background.
Another objection is that many toxicologists do not want to improve toxicity testing. Those in the pharmaceutical industry claim that they are already discarding too many useful compounds. Better toxicity tests would result in even more compounds being rejected. However, in this case they are probably out of touch with the drug developers. Poor toxicity testing results in too many drugs going forward to human clinical trials, which are extremely expensive, only to be rejected on safety grounds. It is this problem which has been noted by the FDA. In testing environmental chemicals more powerful experiments would result in lower NOAELs. This would not be popular with the chemical industry, which probably would object to such improved methods. However, there is currently a 100-fold safety margin for such chemicals, comprising a 10-fold margin for extrapolation to humans and a 10-fold margin to take account of human variation (it is interesting that there is no safety margin to take account of genetic variation in the test animals!). Presumably more accurate toxicity testing, by removing some of the variability and uncertainty, should mean that the safety margin could be reduced. Thus the chemical industry would not necessarily lose out.
The possibility of using a multi-strain experimental design in toxicity testing is now beginning to be taken more seriously by some toxicologists. In June 2005 the NTP held a workshop to discuss a paper that I wrote in 1995 criticising the NTP carcinogenesis bioassay for using only a single strain of mice and rats(13). Geneticists, statisticians and toxicologists discussed the proposals for two days, and the overwhelming conclusion was that a design involving about four isogenic strains chosen from a battery of about 12 strains specifically chosen for toxicity testing would be more powerful and informative than the current design. I understand that the NIEHS is now seriously interested in such experimental designs. This is the only conference that I am aware of where toxicologists actually asked geneticists and statisticians what sort of animals they should use. The answer that they got was that what they are doing at the moment is not scientifically justified.
The NIEHS is also currently funding the full DNA sequence of fifteen mouse strains. Toxicologists are particularly keen on historical data. Having strains of known genotype, and being able to identify DNA polymorphisms will provide a vast pool of historical data because knowledge of the properties of each strain can be accumulated in a way which is not possible when using outbred stocks or a single inbred strain. These strains are being extensively phenotyped for behaviour, spontaneous disease and physiological and immunological characteristics etc., so should become exceptionally powerful tools in toxicity testing (http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=docs/home). When several strains are being used it would also be possible for some of them to be transgenic strains, such as various P450 knockouts, or humanized mice of various types. The multi-strain experiment opens up a wide range of possibilities.
A committee of the US National Academy of Science under the chairmanship of Dr. Daniel Krewski is currently considering the future of toxicity testing of environmental chemicals (http://www8.nationalacademies.org/cp/projectview.aspx?key=74). I gave written and oral evidence to them in July 2005. In thanking me they stated “Your presentation on experimental design and genetic modeling was of great assistance to the committee and generated much discussion in their closed session. The committee was very interested in this topic….â€. While it is not possible to anticipate their conclusions (their report is currently being drafted), I understand that my proposals will receive some support..
Conclusions
According to the FDA, current methods of toxicity testing have changed little over several decades, they are inaccurate, and they have failed to benefit from modern scientific developments. One major factor holding back toxicology is the use of the wrong type of rodents. The proposals given here suggest a way forward which should merit more detailed discussion.
Reference List
- Arcos,J.C., Argus,M.F., and Wolf,G. (1968): Chemical induction of cancer. Academic Press, London.
2. Chia,R., Achilli,F., Festing,M.F., and Fisher,E.M. (2005): The origins and uses of mouse outbred stocks. Nat.Genet., 37:1181-1186.
3. Felton,R.P. and Gaylor,D.W. (1989): Multistrain experiments for screening toxi substances. Journal of Toxicology and Environmental Health, 26:399-411.
4. Festing,M.F. (1986): The case for isogenic strains in toxicological screening. Arch.Toxicol.Suppl, 9:127-137.
5. Festing,M.F. (1990): Use of genetically heterogeneous rats and mice in toxicological research: a personal perspective. Toxicol.Appl.Pharmacol., 102:197-204.
6. Festing,M.F. (1997): Fat rats and carcinogenesis screening. Nature, 388:321-322.
7. Festing,M.F. (2001): Experimental approaches to the determination of genetic variability. Toxicol.Lett., 120:293-300.
8. Festing,M.F.W. (1975): A case for using inbred strains of laboratory animals in evaluating the safety of drugs. Food and Cosmetics Toxicology, 13:369-375.
9. Festing,M.F.W. (1979): Properties of inbred strains and outbred stocks with special reference to toxicity testing. Journal of Toxicology and Environmental Health, 5:53-68.
10. Festing,M.F.W. (1980): Inbred strains and the factorial experimental design in toxicological screening. In: Animal Quality and Models in Biomedical Research. Proceeding of the 7th. ICLAS Symposium, Utrecht, edited by A.Spiegel, pp. 59-66. Gustav Fischer, Stuttgart.
11. Festing,M.F.W. (1987): Genetic factors in toxicology: implications for toxicological screening. CRC Critical Reviews in Toxicology, 18:1-26.
12. Festing,M.F.W. (1993): Genetic variation in outbred rats and mice and its implications for toxicological screening. Journal of Experimental Animal Science, 35:210-220.
13. Festing,M.F.W. (1995): Use of a multi-strain assay could improve the NTP carcinogenesis bioassay program. Environmental Health Perspectives, 103:44-52.
14. Festing,M.F.W. (1997): Variation and its implications for the design of experiments in toxicological research. Comparative Haematology International, 7:202-207.
15. Festing,M.F.W. (1999): Warning: the use of genetically heterogeneous mice may seriously damage your research. Neurobiology of Aging, 20:237-244.
16. Festing,M.F.W., Diamanti,P., and Turton,J.A. (2001): Strain differences in haematological response to chloramphenicol succinate in mice: implications for toxicological research. Food and Chemical Toxicology, 39:375-383.
17. Festing,M.F.W. and Lovell,D.P. (1996): Reducing the use of laboratory-animals in toxicological research and testing by better experimental-design. Journal of the Royal Statistical Society Series B-Methodological, 58:127-140.
18. Festing,M.F.W., Yang,A., and Malkinson,A.M. (1994): At least four genes and sex are associated with susceptibility to urethane-induced pulmonary adenomas in mice. Genetical Research, 64:99-106.
19. Haseman,J.K. and Hoel,D.G. (1979): Statistical design of toxicity assays: role of genetic structure of test animal population. Journal of Toxicology and Environmental Health, 5:89-101.
20. Kacew,S. and Festing,M.F.W. (1996): Role of rat strain in the differential sensitivity to pharmaceutical agents and naturally occurring substances. Journal of Toxicology and Environmental Health, 47:1-30.
21. Pohjanvirta,R., Viluksela,M., Tuomisto,J.T., Unkila,M., Karasinska,J., Franc,M.A., Holowenko,M., Giannone,J.V., Harper,P.A., Tuomisto,J., and Okey,A.B. (1999): Physicochemical differences in the AH receptors of the most TCDD- susceptible and the most TCDD-resistant rat strains. Toxicology and Applied Pharmacology, 155:82-95.
22. Rader,K. (2004): Making Mice. Princeton University Press, Princeton and Oxford.
23. Shellabarger,C.J., Stone,J.P., and Holtzman,S. (1978): Rat differences in mammary tumor induction with estrogen and neutron irradiation. Journal of the National Cancer Institute, 61:1505-1508.
24. Shirai,T., Nakamura,A., Fukushima,S., Yamamoto,A., Tada,M., and Ito,N. (1990): Different carcinogenic responses in a variety of organs, including the prostate, of five different rat strains given 3,2′-dimethyl-4-aminobiphenyl. Carcinogenesis, 11:793-797.